
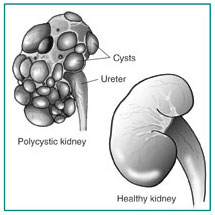
Autosomal Dominant Polycystic Kidney Disease
Abstract – Polycystic kidney disease (PKD) is the commonest life-threatening genetic disease, affecting 12.5 million people worldwide. It is found in all races and occurs equally in men and women. PKD is characterized by the growth of numerous fluid-filled cysts that can profoundly enlarge while replacing much of the normal renal structure, resulting in reduced kidney function and subsequently to renal failure. 1
Two major inherited forms of PKD:
The adult type of PKD has an autosomal dominant type of inheritance (aka ADPKD) and accounts for approximately 90% of all PKD cases. Symptoms usually develop between the ages of 30 and 40, but they can begin earlier, even in childhood.
The less common childhood form of PKD is characterised by an autosomal recessive pattern of inheritance (aka ARPKD). Symptoms of ARPKD begin in the earliest months of life, even in the womb.2
Autosomal Dominant PKD
ADPKD is a multi-systemic and progressive disorder characterized by cyst formation and enlargement in the kidney and other organs. Up to 50% of patients with ADPKD require renal replacement therapy by 60 years of age.
A number of conditions are well-recognised as being associated with ADPKD:
o found in 6% of patients with ADPKD without a family history of aneurysms
o found in up to 16% of patients with ADPKD with a family history
· intracranial dolichoectasia: 2-3% of ADPKD cases
· hypertension: up to 80% of ADPKD cases
· diverticular disease
· valvular heart disease eg. bicuspid aortic valve or mitral valve prolapse (up to 25% of ADPKD cases)
· cysts in other organs including: liver, spleen, pancreas, ovaries, seminal vesicles and prostate.
Symptoms
Clinical presentation is variable and includes:
· dull flank pain of variable severity and time course
· abdominal / flank masses
· haematuria or recurrent urinary tract infections
· hypertension which usually develops at the same time as renal failure
· renal functional impairment to renal failure.
Other symptomatology can arise when above-mentioned complications arise.3
Diagnosis
1. Imaging studies most commonly with ultrasonography; CT or MRI scans are also widely used.
2. Genetic testing is used to detect mutations in one of two genes: PKD1 or PKD2.4
PKD1 is located on chromosome 16p and is responsible for 85% of cases. It is associated with earlier presentation and is more likely to progress to ESRD.
PKD2 is located on chromosome 4q, is responsible for 15% of cases of ADPKD and is typically less severe.
A third rare type of ADPKD (termed ADPKD 3) has been described, however the gene has yet to be identified.5
Other investigations include:
· Standardized blood pressure screening as per American Heart Association recommendations.
· Measurement of blood lipid concentrations because hyperlipidemia is a correctable risk factor for progressive renal disease.
· Urine studies to detect the presence of microalbuminuria/proteinuria, which in the presence of severe renal cystic disease, indicates an increased likelihood of disease progression and mandates strict control of the blood pressure.
· Echocardiography to diagnose valvular heart disease.
· Echocardiography or cardiac MRI to screen patients at high risk because of a family history of thoracic aortic dissections.
· Head MRI or CT angiography to screen patients for intra-cerebral aneurysms.4
Diagnostic criteria
For patients from families with ADPKD of unknown genotype, the following is sufficient for making the diagnosis:
i. in individuals between 15 and 39 years of age, the presence of ≥3 unilateral/bilateral kidney cysts;
ii. in individuals between 40 and 59 years of age, ≥2 cysts in each kidney;
iii. in individuals ≥60 years of age, ≥ 4 cysts in each kidney;
iv. at least 2 cysts in each kidney by age 30 in a patient with a family history of ADPKD can confirm the diagnosis. If there is any question about the diagnosis, a family history of ADPKD and cysts found in other organs make the diagnosis more likely.6
Management
Current therapy is directed towards reducing morbidity and mortality from the renal and extra-renal complications.
Hypertension The antihypertensive agent(s) of choice have not been clearly established. Because of the role of the renin-angiotensin system in the pathogenesis of hypertension in ADPKD, ACE inhibitors and angiotensin II receptor antagonists may be superior to other agents in individuals with preserved renal function.7
Flank pain After excluding causes of flank pain that may require intervention, such as infection, stone, or tumor, an initial conservative approach to pain management is best:
· Nonopioid agents are preferred and care should be taken to avoid long-term administration of nephrotoxic agents such as analgesic and nonsteroidal anti-inflammatory drug combination.
· Tricyclic antidepressants.
· Narcotic analgesics should be reserved for the management of acute episodes.
· Splanchnic nerve blockade with local anesthetics/steroids.
When conservative measures fail, therapy can be directed toward cyst decompression with cyst aspiration and sclerosis. In individuals with many cysts contributing to pain, surgical interventions can be considered including:
· Laparoscopic or surgical cyst fenestration through lumbotomy or flank incision,
· Renal denervation
· In those who have reached ESRD – nephrectomy.8
Cyst hemorrhage and gross hematuria – usually self-limiting and respond well to conservative management with bed rest, analgesics, and adequate hydration to prevent development of obstructing clots.
Rarely, episodes of bleeding are severe with extensive subcapsular or retroperitoneal hematoma, significant drop in hematocrit, and hemodynamic instability. In such cases, individuals require hospitalization, transfusion, and investigation by CT or angiography. In cases of unusually severe or persistent hemorrhage, segmental arterial embolization can be successful. If not, surgery may be required to control bleeding.8
Nephrolithiasis – The treatment of nephrolithiasis in individuals with ADPKD is the same as that for individuals without ADPKD:
· High fluid intake and potassium citrate are the treatment of choice in uric acid lithiasis, hypocitraturic calcium oxalate nephrolithiasis, and distal acidification defects.
· urine alkalinization (to maintain a pH of 6-6.5), and administration of allopurinol.
· Extracorporeal shock-wave lithotripsy and percutaneous nephrostolithotomy.4
Cyst infection – often difficult to treat and has a high treatment failure rate despite prolonged therapy with an antibiotic to which the organism is susceptible. Treatment failure results from the inability of certain antibiotics to penetrate the cyst epithelium successfully and achieve therapeutic concentrations within the cyst. Therapeutic agents of choice include trimethoprim-sulfamethoxazole and fluoroquinolones. Clindamycin, vancomycin, and metronidazole are also able to penetrate cysts well. Chloramphenicol has shown therapeutic efficacy in otherwise refractory disease.9
Malignancy – The diagnosis of renal cell carcinoma in a PKD patient requires a high index of suspicion. MRI with gadolinium enhancement is particularly helpful to detect atypical solid or cystic masses, tumor thrombi, and regional lymphadenopathy.
The diagnosis of transitional cell carcinoma in a polycystic kidney is equally challenging and usually requires retrograde pyelography or ureteroscopy.
End-stage renal disease – Therapeutic interventions aimed at slowing the progression of ESRD in ADPKD include control of hypertension and hyperlipidemia, dietary protein restriction, control of acidosis, and prevention of hyperphosphatemia.
Actuarial data indicate that individuals with ADPKD do better on dialysis than individuals with ESRD from other causes. Females appear to do better than males. The reason for this improved outcome is unclear but may relate to better-maintained haemoglobin levels through higher endogenous erythropoietin production.10
Surveillance
Intracranial aneurysms – Widespread screening is neither cost-effective nor indicated because most intracranial aneurysms found by screening asymptomatic individuals are small, have a low risk of rupture, and require no treatment.
Indications for screening in 20- to 50-year-olds with a good life expectancy include a family history of intracranial aneurysms or subarachnoid hemorrhage, previous rupture of an aneurysm, preparation for elective surgery with potential hemodynamic instability, high-risk occupations such as airplane pilots, and significant anxiety on the part of the individual despite adequate risk information.
Magnetic resonance angiography is the diagnostic imaging modality of choice for presymptomatic screening. Schrier et al recommended rescreening after an interval of ten years because one of 76 individuals with an initial negative study had a new intracranial aneurysm after a mean follow-up of 9.8 years.11,12
Aortic dissection – Until more information becomes available, it is reasonable to screen first-degree adult relatives of individuals with thoracic aortic dissection using either echocardiography or MRI. If aortic root dilatation is found, yearly follow-up and strict blood pressure control with beta blockers should be recommended.
Agents/Circumstances to Avoid
· Long-term administration of nephrotoxic agents such as analgesic and NSAID combinations.
· Caffeine because it interferes with the breakdown of cAMP and hence may promote renal cyst growth.
· Use of estrogens in individuals with severe polycystic liver disease.
· Smoking.
Ongoing Research
Several promising candidate drugs have been suggested including:
ü Tolvaptan, a vasopressin type 2 receptor antagonist – a 5-year multicentre trial which was concluded in 2012 showed that Tolvaptan, as compared with placebo, slowed the increase in total kidney volume and the decline in kidney function over a 3-year period in patients with ADPKD. However, it was associated with a higher discontinuation rate, owing to adverse events.13
ü Octreotide, a somatostatin analogue, has been shown to inhibit kidney growth in a small double-blind, placebo-controlled study done in 2010.14 A phase 3 clinical trial investigating the efficacy of Octreotide in slowing or even halting the kidney enlargement and renal function decline in ADPKD patients with moderate/severe renal failure, is underway (ALADIN 2 study).
ü Calcineurin inhibitors – initial studies had shown that treating adults with ADPKD and early chronic kidney disease with sirolimus did not halt polycystic kidney growth.15 On the other hand, when compared with placebo over a 2-year study period, everolimus slowed the increase in total kidney volume of patients with ADPKD but did not slow the progression of renal impairment.16 Further clinical trials on the efficacy and long-term safety of sirolimus and everolimus are underway.
References
1. Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med 2009; 60:321.
2. Torres VE, Grantham JJ. Cystic diseases of the kidney. In: Brenner BM, ed. Brenner and Rector’s the Kidney. 8th ed. Philadelphia, Pa: Saunders Elsevier; 2007:chap 41.
4. Chapman AB, Rahbari-Oskoui FF, Bennett WM. Course and Treatment of autosomal dominant polycystic kidney disease. UpToDate
5. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007; 369:1287.
10. Grantham JJ, Nair V, Winklhoffer F. Cystic diseases of the kidney. In: Brenner BM, ed. Brenner & Rector’s The Kidney. Vol 2. 6th ed. Philadelphia: WB Saunders Company; 2000: 1699-1730.
11. Schrier RW, Belz MM, Johnson AM et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; Apr: 15(4): 1023-8.
12. Irazabal MV, Huston J 3rd, Kubly V, Rossetti S, Sundsbak JL, Hogan MC et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2011; 6:1274.
13. Torres VE, Chapman AB, Devuyst O et al. Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. NEJM 2012; 367:2407-2418.
14. Hogan MC, Masyuk TV, Page LJ et al. Randomized Clinical Trial of Long-Acting Somatostatin for Autosomal Dominant Polycystic Kidney and Liver Disease. J Am Soc Nephrol, 2010: 10.1681.
15. Serra AL, Poster D, Kistler AD et al, M.D. Sirolimus and Kidney Growth in Autosomal Dominant Polycystic Kidney Disease. N Engl J Med 2010; 363:820-829.
16. Walz G, Budde K, Mannaa M et al. Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N Engl J Med 2010; 363:830-840: 10.1056