
Epigenetic-based treatment as a potential strategy to treat Friedreich’s ataxia
Alfred Grech & Alexandra Baldacchino
Friedreich’s ataxia (FRDA) is an autosomal recessive neurodegenerative disorder. It is the most common inherited ataxia in Europe. The neurodegeneration is progressive and normally, within 15-20 years, the patient becomes wheel-chair bound, and ultimately, is totally incapacitated. Affected individuals normally succumb from heart complications. Frataxin (FXN) is a mitochondrial protein that is deficient in FRDA. The deficiency is caused by a mutation within the first intron of the FXN gene which codes for frataxin. Epigenetic mechanisms are responsible in FRDA and epigenetic-based treatment is a potential new strategy to treat FRDA.
Friedreich’s Ataxia is a Neurodegenerative Disease
Friedreich’s ataxia (FRDA) is an autosomal recessive neurodegenerative disorder. The progressive neurodegeneration, mostly affecting spinocerebellar regions, is due to a deficiency of the protein frataxin.1 Affected individuals suffer from gait disturbances, loss of hand coordination, dysarthria, loss of sensation and loss of muscle power. Diabetes and hypertrophic cardiomyopathy also occur.
Generally, onset occurs before the age of 25 years, but in atypical cases, onset may be delayed or even occur in childhood.1 Normally, within 15-20 years, the patient becomes wheel-chair bound, and ultimately, is totally incapacitated. Affected individuals normally succumb from heart complications.
FRDA is the most common inherited ataxia in Europe and its prevalence is ~ 1 in 30,000.2
Frataxin
Frataxin is the mitochondrial protein which is deficient in FRDA. Controversy still surrounds its function. However, frataxin is believed to be involved in iron homeostasis inside the cell, acting as an iron chaperone and promoting the biogenesis of haem and iron-sulfur clusters (ISCs) inside the mitochondria.3
ISCs act as prosthetic groups for several enzymes and molecules,4 including some that are used inside the mitochondria itself. Thus, frataxin deficiency results in cellular deficiency of these iron-containing enzymes and proteins, mitochondrial dysfunction (decreased Mitochondrial Respiratory Complex I, II and III function) and oxidative damage,5 destining the cell to death.
The Genetic Mutation in FRDA
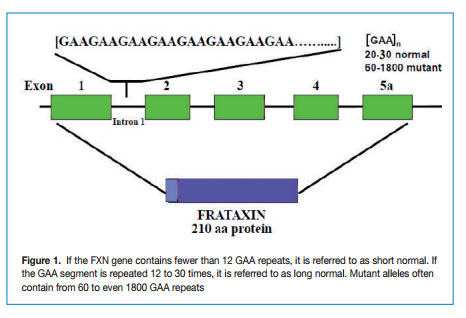
FRDA is caused by a GAA repeat hyper-expansion mutation within the first intron of the FXN gene that codes for frataxin. The cytogenetic location of the frataxin gene is 9q13-q21.1. The difference between normal and affected individuals lies in the number of the GAA repeat sequences; normal alleles contain less than 30 triplets whereas affected alleles have from ~ 60 to more than 1000 GAA triplets (Figure 1).
The Epigenetic Mechanisms Responsible in FRDA
Mounting research is showing that the molecular mechanism could be an epigenetic silencing of the mutated FXN gene. The chromatin in which the gene lies undergoes epigenetic heterochromatisation, i.e. epigenetic mechanisms cause the chromatin to assume a condensed structure and hence it cannot be transcribed because the transcription machinery is prohibited access to the DNA sequence. Characteristically, the heterochromatisation is brought about by classic epigenetic mechanisms, notably DNA methylation and nucleosomal histone modifications. But other epigenetic molecular players may also have a role, e.g. heterochromatin-protein 1 (HP-1), CTCF (CCCTC-binding factor) and FAST-1 (FXN Antisense Transcript-1). The epigenetic aberrations (also called epimutations) that are commonly found in FRDA are hypoacetylation of the nucleosomal core histones H3 and H4 and tri-methylation of the amino acid Lysine in position 9 of histone H3 (H3 Lys9m3). These epimutations involving histones, together with DNA hypermethylation, are very typical of heterochromatin-mediated gene silencing that have also been found in other medical conditions, like cancer (here mostly localised to promoters of tumour suppressor genes).
Epigenetic-Based Treatment as a Potential Strategy to Treat FRDA
Definitely, the FXN gene is an excellent target for epigenetic modulation therapy6 for three valid reasons. First, the most common type of mutation (affecting both alleles) is present in 98% of individuals affected by FRDA. Thus, targeting the epimutated FXN gene is very rational. Secondly, the mutation affects the first intron of the FXN gene. Thus, if the epigenetic aberrations are lifted through epigenetic modulation intervention, it would leave the FXN coding DNA sequence unaffected; transcription followed by splicing would occur and a functioning frataxin protein would be synthesised. Lastly, the heterozygous carriers show no obvious symptoms even though they have ~ 50% deficiency of frataxin. This implies that a good epigenetic-based treatment only needs to increase the frataxin level to modestly approximate that found in carriers.
To address the epigenetic modulation targeting of the FXN gene, several in vitro and in vivo models have been devised. These models, besides serving to study the molecular epigenetic hallmarks associated with the mutated FXN gene (see above), are also being used in the search of new epigenetic-based treatments for the disease. Forexample, Soragni et al.6 created a molecular model of FRDA that could be used to carry out high-throughput analyses of potential substances. They also showed that the GAA*TTC-induced transcriptional silencing in their model could be somewhat alleviated by compounds that have already been shown to stimulate FXN expression in human cell lines.
Herman et al.7 managed to synthesise a class of histone deacetylase inhibitors (HDACIs) that reversed FXN silencing in lymphoblastoid cells and in primary non-replicating lymphocytes from individuals with FRDA. These HDACIs increased the level of frataxin to those of carriers, and no toxicity was observed. The HDACIs directly affected the histones associated with FXN and increased acetylation at particular lysine residues. This class of HDACIs were analogous to the compound BML- 210. Amongst them, the highly active compound was 4b. A follow-up paper by Rai et al.8 , with almost the same researchers as in the paper by Herman et al.7 , made a further step to assess these compounds as potential drugs to treat FRDA. They investigated their efficacy and acute toxicity in an FRDA mouse model (KIKI mouse). One compound was very promising. This was compound 106, a derivative of 4b. This investigation is just an early attempt of epigeneticbased treatment for FRDA. It looks promising but the effect of increased histone acetylation was only evident in the brain for 24 hours after the last injection and had completely disappeared after a week. The KIKI mice just received three subcutaneous doses of compound 106, i.e. once a day for three consecutive days. From this investigation it is clear that further research is needed to see what the correct doses are, in order to maintain the histone modulation continually, without collateral effects (like toxicity) with long term treatments.
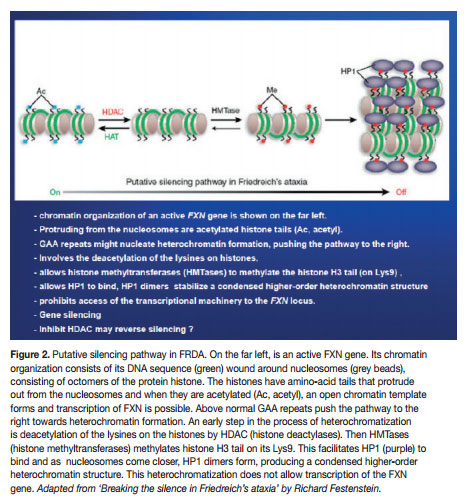
Even though, Soragni et al.6 proposed that their cell line could be adopted for high-throughput analyses in the search for new therapeutics for FRDA, they knew that an appropriate cellular model should not differ greatly from the cell types primarily affected in FRDA, i.e. neurons and heart cells. It was not long that Ku and Soragni et al.9 found the answer in FRDA iPS (induced pluripotent stem) cells. They reprogrammed fibroblasts into induced pluripotent stem cells which were then differentiated into neurons. They then demonstrated that the FRDA neuronal cells responded to HDACIs. In 2011, permission was given to Repligen Corporation of Waltham, US, for pre-clinical investigation of candidate (RG2833), and Phase I safety trials in human subjects are on their way.10 HDACIs are surely showing superiority. But epigenetic modulation can also be made through other molecular players. Indeed, translational therapeutic research should also be done with histone methylation reducing drugs (e.g. splitomicin), DNA methylation inhibitors (e.g. zebularine) and GAA-interacting compounds (e.g. pentamidine and DB221).
Conclusion
Epigenetic-based treatment strategies are rational and the first generation of epigenetic-based drugs has already been approved by FDA, e.g. for myelodysplastic syndrome. Indeed, the use of such drugs is confirming that epigenetic modulation can be a feasible treatment option, not only for cancer, but also for a growing list of diseases where epigenetic mechanisms of gene expression underline their pathogenesis.11 Since epigenetic changes are potentially reversible and are thought to be responsible for a wide range of diseases, the scope of epigenetic therapy is likely to expand.12 And FRDA is surely next in line.
References
1. Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews [Internet]. University of Washington, Seattle; 1993-2013. Available from: www.ncbi. nlm.nih.gov/books/NBK1116/
2. Delatycki MB, Williamson R, Forrest SM. Friedreich ataxia: an overview, J Med Genet 2000; 37:1-8.
3. Chen OS, Hemenway S, Kaplan J. Inhibition of Fe-S cluster biosynthesis decreases mitochondrial iron export: evidence that Yfh1p affects Fe-S cluster synthesis. Proc Natl Acad Sci 2002; 99:12321-6.
4. Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet 1997; 17:215-7.
5. Pandolfo M. Frataxin deficiency and mitochondrial dysfunction. Mitochondrion 2002; 2:87-93.
6. Soragni E, Herman D, Dent SY et al. Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia. Nucleic Acids Res 2008; 36(19):6056-65.
7. Herman D, Jenssen K, Burnett R et al. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006; 2:551-8.
8. Rai M, Soragni E, Chou CJ et al. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich’s ataxia patients and in a mouse model. PLoS One 2010; 5(1):e8825.
9. Ku S, Soragni E, Campau E et al. Friedreich’s Ataxia Induced Pluripotent Stem Cells Model Intergenerational GAA*TTC Triplet Repeat Instability. Cell Stem Cell 2010; 7(5):631-7.
10. Gottesfeld J. Development of histone deacetylase inhibitors as therapeutics for Friedreich’s ataxia. 2011. Available at: www. ataxia.org/events/2011-Presentations/Saturday/ NAFGottesfeld031911.pdf
11. EpiGenie. Epigenetic Drug Therapies On The Rise. 2011. Available at: http://epigenie.com/
12. Peedicayil J. Epigenetic Therapy-A New Development In Pharmacology. Indian J Med Res 2006; 123:17-24.