
Oncogene addiction might be the Achilles heel in cancer – Part II
by Alfred Grech & Alexandra Baldacchino
Molecular mechanisms of oncogene addiction
Various hypotheses have been put forward to account for the molecular mechanism of oncogene addiction, with the main ones being synthetic lethality and oncogenic shock. A favoured hypothesis is that of synthetic lethality. Two genes are said to be synthetically lethal if mutation of one of the genes is compatibale with cell survival but mutation in both genes results in cell death. It has been suggested that in a cancer cell, the activating oncogene is in a synthetic lethal relationship with a gene that is inactivated in the cancer cell. Accordingly, eliminating the oncogene would lead to cancer cell death32,33 while sparing normal cells. Using this fact to their advantage, Puyol et al.34 unveiled a therapeutic strategy for non-small cell lung carcinoma after discovering that a synthetic lethal interaction exists between K-Ras oncogenes and Cdk4.
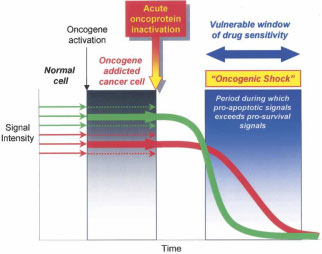
One other favourite mechanism that has also been put forward to explain oncogene addiction is referred to as ‘oncogenic shock’. Here, Sharma et al.35 proposed that the acute inactivation of the oncogene protein (oncoprotein), results in a ‘differential decay rates of various prosurvival and proapoptotic signals’ associated with the oncoprotein (figure 1). Indeed, they suggested that the prosurvival signals (green arrows) are transient and dissipate relatively quickly upon oncogene inactivation, whereas the proapoptotic signals (red arrows) linger for a longer period of time thus committing the tumour cell to apoptotic death.
Figure 1. Relationship between oncogene addiction and oncogenic shock In an oncogene addicted cancer cell, the prosurvival signals (green arrows) predominate over the proapoptotic signals (red arrows) and result in the survival of the cancer cell. Following acute oncoprotein inactivation, prosurvival signals dissipate rather quickly relative to the proapoptotic signals which are prolonged. Thus the lingering proapoptotic signals cause the cells to irreversibly undergo apoptosis (Source: Sharma and Settleman, 200736).
Consistent with this model, Sharma et al.35 observed that when lung cancer cells are treated with gefitinib, which, like erlotinib, is an epidermal growth factor receptor (EGFR) inhibitor, they are more efficiently killed than when they are treated with the prosurvival receptor ligand, EGF.
Integrating new approaches into the clinical setting in order to characterise the state of oncogene addiction
Identifying the particular state of oncogene addiction in specific types of human cancer can be conducive to treating patients with appropriate molecular agents. Presently, there is no way to fully assess the total signalling pathways, inside and outside normal or cancer cells, that control their proliferation, differentiation and apoptosis. However, advances are being made in profiling patterns of gene expression, genomics, epigenomics, proteomics, network theory, systems biology and computer modelling. These advances will eventually help in identifying the Achilles’ heel in specific types (and their subtypes) of human cancer. Integrating all these techniques would then lead to tailor-make optimal therapy by developing agents that target the critical oncogene. Also they would become useful in ‘oncogenic escape’ states.
Cancers can ‘escape’ from a particular state of oncogene addiction. This results due to mutations in other genes and pathways, probably because of the genomic instability of cancers. Moreover, many research papers, such as that by Giuriato and Felsher,37 even suggest that upon sustained oncogene inactivation, some cancers relapse and are thus no longer dependent on the oncogene to which they were previously addicted to. This explains why using single molecular targeted agents may not achieve long-lasting remissions or cures and one needs to opt for combination therapies in such situtations. But again combination therapies should be rationally designed using the integrative approaches mentioned above.
Currently, choosing the best molecular targeted agent, alone or in combination, for a specific patient with cancer is largely empirical. But this scenario is rapidly changing, as the oncologist can now choose from a rapidly developing list of diverse molecular targeted agents. This coupled with several research mechanistic studies and techniques to profile the molecular networks in human cancer and their subtypes, should exploit the concept of oncogene addiction and be conducive to more rational, effective and tailor-made therapies for cancer.
Conclusion
Cancer is multifaceted, involving many interactions between different genes, pathways and signalling cascades. This makes the detection of a single marker molecule, and thus the determination of oncogene addiction, rather complex. Besides, it has also been reported by Tonon38 that genetic abnormalities in cancers tend to gather around specific pathways, giving way to the concept of ‘network addiction’, rather than oncogene addiction. Therefore, the development of new integrative strategies for defining these oncogene addiction networks together with the use of molecular target agents, might, in the near future, make it possible to achieve more effective, tailor-made therapies for the treatment of human cancer.
References
1. Weinstein IB, Begemann M, Zhou P, Han EK, Sgambato A, Doki Y et al. Disorders in cell circuitry associated with multistage carcinogenesis: exploitable targets for cancer prevention and therapy. Clin Cancer Res 1997; 3:2696-702.
2. Weinstein IB. Cancer. Addiction to oncogenes-the Achilles heal of cancer. Science 2002; 297:63-4.
3. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314:268-74.
4. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153-8.
5. Arber N, Doki Y, Han EK, Sgambato A, Zhou P, Kim NH et al. Antisense to cyclin D1 inhibits the growth and tumourigenicity of human colon cancer cells. Cancer Res 1997; 57:1569-74.
6. Kornmann M, Danenberg KD, Arber N, Beger HG, Danenberg PV, Korc M. Inhibition of cyclin D1 expression in human pancreatic cancer cells is associated with increased chemosensitivity and decreased expression of multiple chemoresistance genes. Cancer Res 1999; 59:3505-11.
7. Li K, Lin SY, Brunicardi FC, Seu P. Use of RNA interference to target cyclin E-overexpressing hepatocellular carcinoma. Cancer Res 2003; 63: 3593-7.
8. Colomer R, Lupu R, Bacus SS, Gelmann EP. erbB-2 antisense oligonucleotides inhibit the proliferation of breast carcinoma cells with erbB-2 oncogene amplification. Br J Cancer 1994; 70:819-25.
9. Verma UN, Surabhi RM, Schmaltieg A, Becerra C, Gaynor RB. Small interfering RNAs directed against beta-catenin inhibit the in vitro and in vivo growth of colon cancer cells. Clin Cancer Res 2003; 9:1291-300.
10. Aoki K, Yoshida T, Matsumoto N, Ide H, Sugimura T, Terada M. Suppression of Ki-ras p21 levels leading to growth inhibition of pancreatic cancer cell lines with Ki-ras mutation but not those without Ki-ras mutation. Mol Carcinog 1997; 20:251-8.
11.Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumourigenicity by virus-mediated RNA interference. Cancer Cell 2002; 2:243-7.
12. Miller AJ, Du J, Rowan S, Hershey CL, Widlund HR, Fisher DE. Transcriptional regulation of the melanoma prognostic marker melastatin (TRPM1) by MITF in melanocytes and melanoma. Cancer Res 2004; 64:509-16.
13. Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumours. Cancer Res 2005; 65:2412-21.
14. Huettner C S, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet 2000; 24:57-60.
15.Felsher DW, Bishop JM. Reversible tumourigenesis by MYC in hematopoietic lineages. Mol Cell 1999; 4:199-207.
16. D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE et al. c-MYC induces mammary tumourigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med 2001; 7:235-9.
17. Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002; 297:102-4.
18. Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002; 109:321-34.
19. Moody SE, Sarkisian CJ, Hahn KT, Gunther EJ, Pickup S, Dugan KD et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell 2002; 2:451-61.
20. Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999; 400: 468-72.
21. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R et al. Analysis of lung tumour initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 2002; 15:3243-8.
22. Gunther EJ, Moody SE, Belka GK, Hahn KT, Innocent N, Dugan KD et al. Impact of p53 loss on reversal and recurrence of conditional Wnt-induced tumourigenesis. Genes Dev 2003; 17:488-501.
23. Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 2003; 349:1423-32
24. Baselga J, Trigo JM, Bourhis J, Tortochaux J, Cortes-Funes H, Hitt R et al. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol 2005; 23:5568-77.
25. Moore MJ. Brief communication: a new combination in the treatment of advanced pancreatic cancer. Semin Oncol 2005; 32:5-6.
26. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005; 353:123-32.
27.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783-92.
28. Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 2005; 353:1659-72.
29. Miller KD, Chap LI, Holmes FA, Cobleigh MA, Marcom PK, Fehrenbacher L et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol 2005; 23:792-9.
30. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350:2335-42.
31. Lugo TG, Pendergast A M, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990; 247:1079-82.
32. Reddy A, Kaelin WG Jr. Using cancer genetics to guide the selection of anticancer drug targets. Curr Opin Pharmacol 2002; 2:366-73.
33. Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5:689-98.
34. Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010; 18:63-73.
35. Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res 2006; 12:4392s-4395s.
36. Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 2007; 21:3214-31.
37. Giuriato S, Felsher DW. How cancers escape their oncogene habit. Cell Cycle 2003; 2:329-32.
38. Tonon G. From oncogene to network addiction: the new frontier of cancer genomics and therapeutics. Future Oncol 2008; 4:569-77.