
Unravelling the Tangle of Genetic Testing – Part III
by Christian A Scerri MD PhD(Molecular Genetics)
Clinical and Molecular Geneticist
Clinical and Molecular Genetics Clinic
Medical School, St Luke’s Hospital
The Clinical Genetic Consultation
The referring physician (either the primary physician or the specialist) are usually the first to suspect the genetic basis of the condition. Though the differential diagnosis would thus have already been performed, this would usually result in either a possible number of suspect genes or a number of possible mutations within a gene and in most conditions, both of these possibilities. Thus the clinical geneticist has to further dissect the signs and symptoms of the disease so as to focus on the most probable candidate gene.
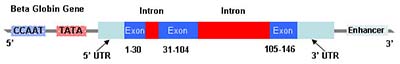
Figure 1: The beta globin gene on chromosome 11
Once a candidate gene has been identified, the next step is to confirm or exclude the presence of mutations within this gene. The initial reaction to this would be that as we now have the whole of the human genome sequenced, then sequencing the whole gene should indicate the presence or otherwise of mutations. This is partly true for small genes (around 2000 base pairs e.g. beta globin – see figure 1) as a full sequencing of the gene would take a reasonably short time at an affordable cost. On the other hand, sequencing large genes (e.g. CFTR – cystic fibrosis; dystrophin – Duchenne and Beckers muscular dystrophies; pyrin gene – familial Mediterranean fever; BRCAI and BRCA II – breast and ovarian cancer) would be both time consuming as well as very expensive. Thus a cheaper and faster strategy has to be devised. Amongst stable populations that lack large and recent migrations (such as Malta), a small (1 to 4) number of mutations would usually account for almost 100% of the mutations within a particular gene of interest. This is particularly true for a number of single gene disorders listed in table 1. Thus, by utilizing techniques (also listed in table 1) that target these particular mutations, one can identify them in a reasonably short time and at a reasonable expense. The utilisation of these techniques requires prior knowledge of the molecular epidemiology of the disease in a particular population (and a full molecular characterization of the disease in the same population).
Unfortunately, in other single gene disorders where de novo mutations are the norm, this method cannot be utilized as no particular mutations would predominate amongst the rest. Typical examples of this group are the Dystrophin gene and the FBN1 gene (protein fibrillin-1) responsible for the Marfan’s syndrome. In these cases the strategy would involve the low resolution screening of the whole gene in the proband, with sequencing of the areas that show a possibility of mutations. Once a mutation is characterised within the family, the specific test could then be utilised to identify this mutation in other family members.
Though these testing schemes seem to be very logical and thus should give a result in every case, this is not always true. A number of actual examples from the Clinical and Molecular Genetics Clinic, at the Medical School, St Luke’s Hospital should demonstrate this.
Case 1: The proband was a 5 year old boy with low grade anaemia, microcytosis, an elevated HbA2 level and a normal body iron level. His mother had a normal blood picture and a normal HbA2 level whilst his father had a mean corpuscular volume at the lowest end of the normal range and an elevated HbA2 level. Full sequencing of the beta-globin gene did not identify any mutations or gross deletions. Thus one was faced with the dilemma of whether this boy was a carrier for beta-thalassaemia or not.
Case 2: The proband was a 14 year old girl with symptoms typical of Familial Mediterranean Fever. From the family history, it was apparent that her father had a similar history but neither her mother nor her 16 year old sibling had any suggestive symptoms. Molecular analysis showed that the proband, her sibling and her father all carried one mutated gene, whilst her mother had no apparent mutations in the pyrin gene. Both proband and father responded well to colchicines treatment. In this case there are three dilemmas – as this is a recessive disorder, what is the most probable explanation for the seemingly dominant picture; should the second sibling be treated even though s/he has no symptoms and what type of counselling in regard to future offspring of both siblings can one give?
Case 3: This involves two brothers that were both clinically suffering from muscular dystrophy but without any previous family history of the condition. Muscular biopsy showed a severe Duchenne type in one and a less severe Becker’s type in the second. Dystrophin gene analysis showed no gross deletions or mutations. The question that arises is how could a presumably identical mutation give rise to two different clinical pictures. And in the absence of a clearly identified mutation, what is the genotype and thus the carrier status of the two, apparently healthy sisters?
From the above it is clear that though genetic testing has the potential of confirming the diagnosis as well as offering the tools by which one can offer effective family counselling, there are a number of occasions (in our experience, estimated at around 20% of cases) where molecular genetic results would either offer no additional information on the disorder or present an even more confusing picture. It is thus imperative that further research on the molecular pathophysiology of these disorders is carried out so as to define the molecular interactions not only in the multifactorial disorders but also in those that are today considered monogenic disease. In addition, molecular epidemiological work amongst the Maltese population is further required so as to identify the particular mutations that are present in the population and thus make their identification possible in the shortest time and in the least expensive way. This requires not only the work of the molecular geneticist but also the co-operation of specialists and primary physicians to identify and refer patients as well as to participate in population-based screening programmes.
Bibliography
Ashtar A. Molecular Pathology of Infantile GM1-ganglioasidosis. Malta: University of Malta, 1998.
Avery OT, Macleod CM, McCarty M. Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types. J Exp Med 1944; 79:137-58. 1944.
Bezzina-Wettinger S, Balim Z, Felice AE. Allele Frequencies of selected polymorphisms related to thrombosis in the Maltese Population. Blood 2000; 96 part 2: 94b.
Farrugia R, Scerri CA, Montalto SA, Parascandolo R, Neville BG, Felice AE. Molecular genetics of tetrahydrobiopterin (BH4) deficiency in the Maltese population. Mol Genet Metab 2007; 90(3):277-83.
Franklin RE, Gosling RG. Evidence for 2-chain helix in crystalline structure of sodium deoxyribonucleate. Nature 1953;172(4369):156-7.
Galdies R, Pullicino E, Cassar W, Bezzina-Wettinger S, Borg J, Felice AE. A first study on the frequency and phenotypic effects of HFE gene mutations in the Maltese population. Malta Medical Journal 2006; 18[Supplement].
Kellogg DA, Doctor BP, Loebel JE, Nirenberg MW. RNA codons and protein synthesis. IX. Synonym codon recognition by multiple species of valine-, alanine-, and methionine-sRNA. Proc Natl Acad Sci USA 1966; 55(4):912-9.
Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 1986;51 Pt 1:263-73.
Scerri CA. Clinical and Molecular Pathology of the beta+ IVSI-6C Thalassaemia in Malta. Malta: University of Malta, 1998.
Vella J. The Detection of the DNA Mutations that Cause Gangliosidosis. Malta: University of Malta, 2000.
Watson JD, Crick FH. The structure of DNA. Cold Spring Harb Symp Quant Biol 1953; 18:123-31.
Wilkins MH, Seeds WE, Stokes AR, Wilson HR. Helical structure of crystalline deoxypentose nucleic acid. Nature 1953;172(4382):759-62.
| Disease | Gene | Size (base pairs) | Frequency
Population |
Mutations | Consequence of Defect | Allele Frequency in Homozygotes | Test Methods | |
| Hetero-zygotes | Homo-zygotes | |||||||
| Beta Thalassaemia | Beta Globin Gene | ~2000 | 1.8%(7) | 0.01% | IVS1-6C | RS | 70.7%(7) | SEQ |
| IVSII-1A | AS | 10.3%(7) | SEQ | |||||
| IVSI-110A | RS | 12.1%(7) | SEQ | |||||
| Cod 39T | PSC | 6.9%(7) | SEQ | |||||
| Familial Mediterranean Fever | Pyrin
Gene |
~15000 | 10.4%(8) | 0.25% | V726A | AAC | 22.0(8) | SEQ |
| M694V | AAC | 19.0(8) | SEQ | |||||
| E148Q# | AAC | — | SEQ | |||||
| Haemo-chromatosis | HFE gene | ~4500 | 31.5%(9) | 3.8% | C282Y | AAC | 0%(9) | RE-D |
| H63D | AAC | 97%(9) | RE-D | |||||
| S65C | AAC | 3%(9) | RE-D | |||||
| Dopamine Responsive Dystonia | SPR Gene | ~5000 | 4.6%(10) | 0.06% | IVS2-2G | AS | 100%(10) | RE-D |
| Atypical Phenoketonuria | QDPR
(DHPR) |
~26000 | 3.3%(10) | 0.03% | G23D | AAC | 100%(10) | RE-D |
| Gangliosidosis | GLB1 | ~100000 | 3%*(11;12) | 0.02%* | R482H | AAC | – | SEQ |
| IVS7-9,10 | RS | – | SEQ | |||||
| IVS14-2G | AS | – | SEQ | |||||
| Factor V Leiden | F5 | ~73000 | 2.6%(13) | – | R506Q | AAC | N/A | RE-D |
| Factor II | F2 | ~20000 | 3%(13) | – | G20210A | 3’ UTR | N/A | RE-D |
| Methylene –tetrahydrofolate Reductase deficiency | MTHFR | ~20000 | 49.6%(13) | 9.1% | C677T | AAC | N/A | RE-D |
| ?† | ?† | A1298C | AAC | N/A | RE-D | |||
| Cystic Fibrosis | CFTR | ~189000 | ?† | ?† | Delta508 | PSC | N/A | ASOH |
| 47 mutations | Various – mostly AAC | N/A | ASOH |
Table 1 Some common single gene disorders present in the Maltese population and that are regularly seen and tested for at the Clinical and Molecular Genetics Clinic at the Medical School, SLH.
RS – Reduced Splicing; AS – Abnormal Splicing; PSC – Premature Stop Codon; AAC – Amino Acid Change; 3’UTR – 3’ Untranslated Region – RNA instability; SEQ – Direct Sequencing; RE-D – Restriction Enzyme Digestion; ASOH – Allele Specific Oligonucleotide hybridisation. Note. In the case of Factor V Leiden and Factor II, the presence of one mutated allele (thus heterozygotes) is associated with an increased risk of thrombophilic episodes. # E148Q mutation was not found in the patient cohort though it was present in 6.4% of the random population tested – most probably due to the mildness of the condition associated with this mutation. *Estimated incidence – no population wide studies have been conducted as yet. † No population wide studies have been conducted so far.
{kind=link}