Thalassaemia – A review
by Christian A Scerri MD PhD(Molecular Genetics)
Clinical and Molecular Geneticist
Clinical and Molecular Genetics Clinic
Speciality Clinics, Mater Dei Hospital
Although reference to a disease causing anaemia in children can be found in ancient Greek and Italian writings1, the first clinical description of thalassaemia as a separate entity, was done by Cooley and Lee in 1925.2 Since then b-thalassaemia has also been known as Cooley’s anaemia. The term ‘thalassaemia’ was in fact coined by Whipple and Bradford3 in 1932, in their paper on the pathology of the disease. Thalassaemia is derived from the Greek word Qalassa (Thalassa) meaning ‘the sea’.
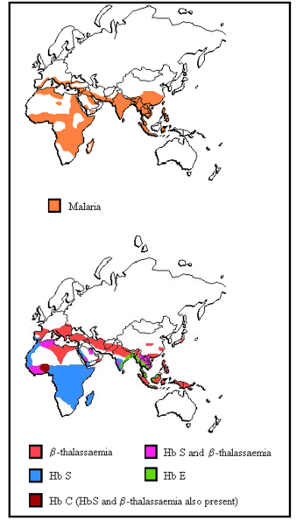
It has been generally accepted that the high incidence of thalassaemia and the common haemoglobin variants, Hb S, Hb E and HB C, in certain areas of the world is the result of selective pressures of Plasmodium falciparum malaria.4,5 As can be seen in figure 1, the distribution of these disorders follow quite closely the distribution of P. falciparum. There is good evidence to believe that carriers of one mutated gene had a higher reproductive fitness in malarial areas as compared to normal individuals, possibly due to an inability of the intracellular parasite to complete its life cycle.
Figure 1 Distribution of malaria and b-thalassaemia, Hb S, Hb C and Hb E
HUMAN HAEMOGLOBINS – STRUCTURE
Human haemoglobins are tetramers of two a-globins, i.e. z or a, and two non-a-globins, i.e. e, Gg, Ag, d, or b, each associated with a haem group. The haem group is formed by an iron atom surrounded by a porphyrin ring. The genes that encode for the globins are arranged 5′ to 3′ in the same order in which they are sequentially expressed during development. Thus the a-globin gene cluster, spanning a region of around 50 kilobases (Kb) on the short arm of chromosome 16, contains the embryonic z gene and two a genes – a2 and a1, together with a number of non-functional pseudo-genes.
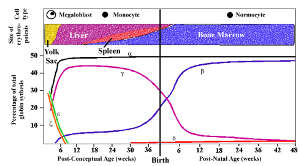
Figure 2. Globin chain synthesis at different stages of foetal maturation (Source: Weatherall DJ and Clegg JB, The Thalassaemia Syndromes2)
The non-a-gene cluster, spanning a region of around 90Kb on the short arm of chromosomes 11, comprises, from 5′ to 3′, the embryonic e-gene, two foetal genes, Gg and Ag, and the adult d and b genes.
MOLECULAR STRUCTURE OF GLOBIN GENES
Being only around 2 Kb long, globin genes are relatively small, in contrast to other genes (blood coagulation Factor VII gene ~13Kb; human dystrophin gene ~2 megabases Mb). The relatively small size facilitated the identification and characterisation of mutations leading to thalassaemia and other haemoglobin variants.
In thalassaemia, there is a reduction of one of the globin chains due to mutations or deletions affecting DNA transcription or translation. a-thalassaemia (affecting a-chain production) and b-thalassaemia (affecting b-chain production) are common and clinically important.
Whilst the majority of β-thalassaemia conditions are due to point mutations involving one or two nucleotides, α-thalassaemia is commonly due to large deletions involving the whole gene. Thalassemia mutations and deletions are relatively population specific and Malta is no exception. Around 72% of the β-thalassaemia cases are due to the relatively mild IVS1-6T->C mutation (i.e. a Cytosine to Thymine substitution in the 6th nucleotide of the first intron sequencing). 6 This mutation decreases the efficiency of normal splicing (to about 30% of normal), resulting in an increase in the utilisation of the alternative splice sites and a prematurely terminated protein. Three other mutations, codon 39 C->T (insertion of a premature stop codon), the IVS-I-110 G->A (production of a new acceptor splice site in the first intron) and the IVS-II-1 G->A (abolition of the splice site), make up the remaining 28%. In the case of α-thalassaemia, a 3.7 Kb deletion (basically this deletion reduces the number of α gene copies to 1 from the normal 2 genes per chromosome 16), is the only α gene deletion discovered amongst the Maltese population.
From the knowledge of the molecular pathology, it is clear that the major problem in thalassaemia is a disbalance between the α and β globins that are an integral part of the heamoglobin molecule. Thus in β-thalassaemia, there is a relatively higher level of α globin in relationship to β-globin and vice versa in α-thalassaemia. The unpaired, excess globin chains either form unstable tetramers (β chains – HbH and γ chains – HbBarts) or else the α chains bind to the erythrocyte membranes causing membrane damage . In both cases, apart from inefficient erythropoiesis there is also increased intra-medullary haemolysis.
Whilst thalassaemia carriers (i.e. carrying one mutated allele in the case of β-thalassaemia and up to 2 mutated/deleted alleles in the case of α-thalassaemia) are usually asymptomatic, they have a slightly lowered haemoglobin level (10-12g/dl) and microcytosis (a MCV lower than 80fl and a MCH lower than 26pg) with a high Hb A2 (higher than 3.5%) in the case of β thalassaemia carriers. The identification of thalassaemia carriers is further complicated by concurrent presence of iron deficiency. In the case of β-thalassaemia, the presence of mutations causing mild disease, or concurrent presence of α-thalassaemia or δ-β thalassaemia (deleterious mutations in both the δ and β genes) can further complicate its diagnosis.
Thalassaemia Major
Thalassaemia major can be divided into α0 or β0 and α+ or β+ depending on the amount of the respective residual globin chain production. Whilst α0 and β0 thalassaemias are present with very severe anaemia, in α+ and β+ thalassaemias the picture is more varied. α+ thalassaemia can be divided into two distinct groups:
1. Individuals that inherit Thalassemia 3 normal α-genes (-α/αα). These individuals are referred to clinically as silent carriers of α-thalassemia or α-thalassemia-2 trait. The affected individuals exhibit no clinical abnormality and usually have mild reductions in red cell mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH).
2. The inheritance of 2 normal alpha genes either due to heterozygosity for alpha (0) thalassemia (αα/–) or homozygosity for alpha (+) thalassemia (-α/-α) results in the development of α-thalassemia minor or α-thalassemia-1 trait. Similarly to α-thalassemia-2, the affected individuals are clinically normal with minimal anaemia and reduced MCV and MCH.
3. HbH disease or the inheritance of one normal alpha gene (-α/–), is the result of abundant formation of hemoglobin H composed of tetramers of excess beta chains. The affected individuals have moderate to severe lifelong haemolytic anemia, modest degrees of ineffective erythropoiesis, splenomegaly and variable bony changes.
The clinical picture of β+ thalassaemia is more varied as it depends on the inherited mutation and other genetic (and probably environmental) factors. The clinical picture can range from a severe to moderate anaemia (Hb levels of 5 to 8 g/dl) to very mild anaemia (9-10g/dl).
Clinical Management of Thalassaemia
The classical management of thalassemia is based on an adequate transfusion regime and proper iron chelation. The transfusion regime (usually every 4-6 weeks) is aimed at ensuring that the mean haemoglobin level is around 9-10g/dl, thus reducing the lifelong complications associated with severe, chronic anaemia. Coupled with the transfusions, these individuals require adequate iron chelation, to reduce iron overload. Up to now, this was achieved through the daily, subcutaneous injection of deferoxamine administered through a syringe pump but newer, oral chelators have been developed and are presently being introduced into the market. These oral chelators are offering both a reduction in the discomfort from the subcutaneous injection as well as generally improving the compliance and thus reducing iron overload complications.
In addition to transfusion and chelation, splenectomy has the potential of reducing the transfusion requirement and is usually performed just before puberty. Furthermore, bone marrow transplant, when appropriate and where a donor is available, offers a cure, as opposed to treatment, for thalassaemia major individuals.
References
1. Bannerman RM. Thalassaemia: A survey of some aspects. New York: Grune and Stratton; 1961.
2. Cooley TB, Lee P. A series of cases of splenomegaly in children with anaemia and peculiar bone changes. Trans Am Pediatr Soc 37: 29-30, 1925.
3. Whipple GH, Bradford WL. Mediterranean disease-thalassaemia (erythroblastic anaemia of Cooley); associated pigment abnormalities simulating hemochromatosis. J Pediatr 1932; 9:279.
4. Weatherall DJ, Clegg JB. The Thalassaemia Syndromes. Oxford: Blackwell Scientific Publications; 1981.
5. Nagel RL, Roth EF. Malaria and red cell genetic defects. Blood 1989; 74:1213-21.
6. Scerri CA. Clinical and Molecular Pathology of the b+ IVSI-6C Thalassaemia in Malta [dissertation]: University of Malta; 1998.